State governments should
-
eliminate government licensing of medical professionals—or, as a preliminary step, recognize licenses from other states and third-party credentialing organizations;
-
eliminate “certificate of need” laws;
-
eliminate price controls, including “parity” laws for telehealth and other services; and
-
direct courts to enforce private contracts in which patients and providers agree on alternative medical malpractice liability rules.
Congress should
-
eliminate states’ ability to use licensing laws as a barrier to entry by medical professionals who hold licenses from other states;
-
eliminate the U.S. Food and Drug Administration (FDA)—or, as preliminary steps,
- eliminate the FDA’s premarket-approval requirements for drugs and medical devices,
- eliminate the FDA’s power to mandate prescriptions for drugs and medical devices, and
- eliminate the FDA’s power to limit truthful speech;
-
recognize drug and device approvals by other third-party organizations, including foreign regulators; and
-
reject federal medical malpractice reforms.
The most important health care right is the right to make one’s own health decisions. When government regulations deny consumers their choice of providers and treatments, or when government refuses to enforce certain contracts, it violates patients’ rights to make their own health decisions and reduces access to care.
Making health care better, more affordable, and more secure, particularly for the most vulnerable, requires restoring those rights. Policymakers must eliminate regulations that deny consumers the right to make their own health decisions and must honor contracts between competent patients and providers.
End Government Licensing of Medical Professionals
Government licensing of clinicians violates the right of patients to choose their providers, makes health care less accessible by increasing prices, and reduces the quality of medical care.
Markets make medical care more affordable in part by allowing competent clinicians with less training than physicians, such as nurse practitioners and physician assistants, to perform progressively more tasks. Markets improve quality in part by allowing clinicians to combine their skills in various ways. Among the quality-improving innovations that markets have produced are integrated group health plans that coordinate care and offer other efficiencies. Patients have a right to choose to receive medical care from independent nurse practitioners, integrated group plans, or any other arrangement entrepreneurs offer.
Clinician licensing blocks entry by these and other providers. It therefore blocks the market processes that make health care better, more affordable, and more secure.
To practice medicine in a state, clinicians—physicians, nurse practitioners, physician assistants, dentists, dental hygienists, and others—must obtain a license from that state. Each state defines which clinician categories may exist. The states mandate minimum educational requirements for each profession. They define the list of tasks, or “scope of practice,” that each license allows members of that profession to perform. States delegate these highly technical decisions to members of the health professions—typically physicians or dentists, who have the greatest understanding of the science of medicine and dentistry.
These are not scientific decisions. If they were, all states would have identical rules. Instead, state licensing laws vary dramatically on whether they allow nurse practitioners to prescribe medication (see Figure 1) or practice independently (see Figure 2), whether they allow dental therapists to practice at all (see Figure 3), and other dimensions of medical and dental practice.
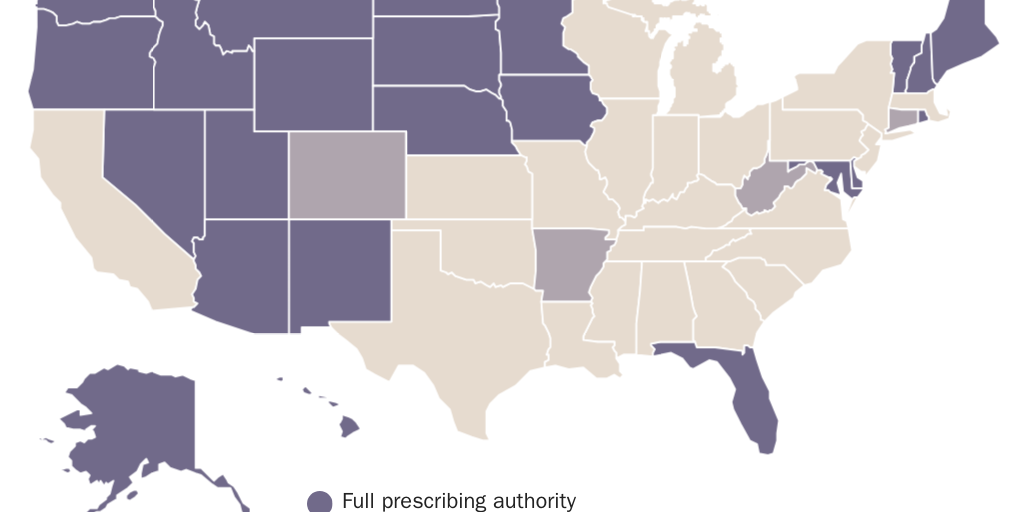
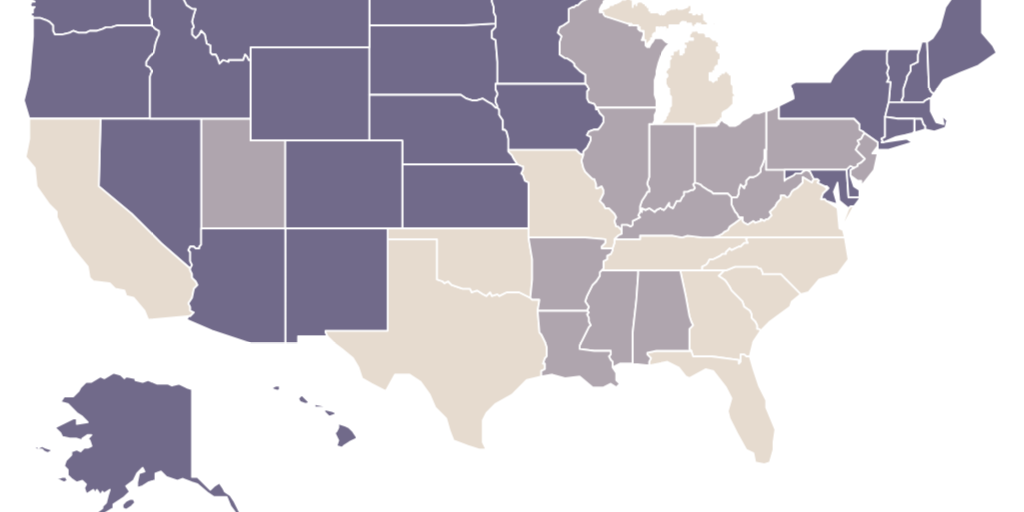
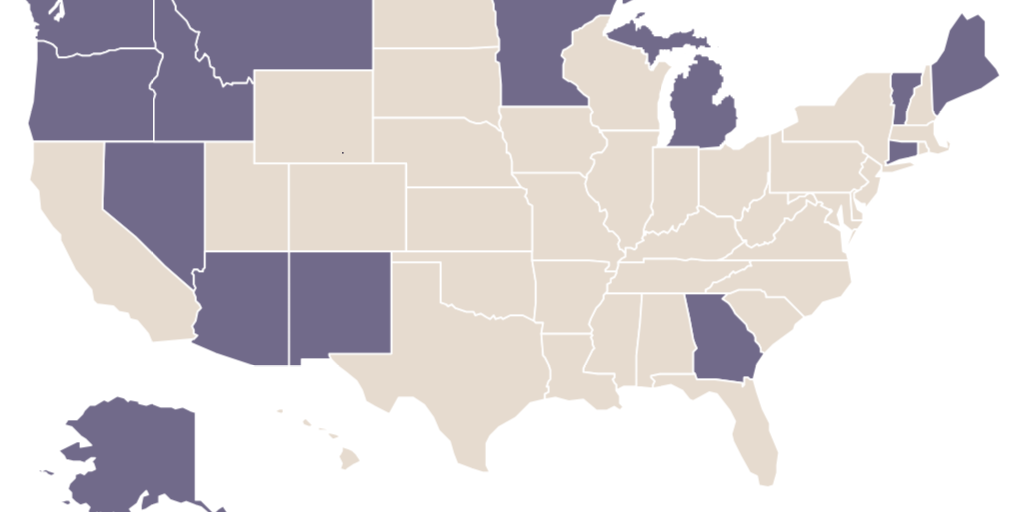
Licensing gives self-interested incumbents—typically, physicians and dentists—the power to set rules for new entrants into their own profession and other health professions. In other words, it empowers incumbent clinicians to create barriers to entry for their competitors.
It should come as no surprise, therefore, that licensing typically leads to “steadily rising requirements” for entry into the health professions and that incumbents use licensing laws to block their competitors from providing particular services. The American Medical Association lobbies on behalf of physicians. It boasts that it has blocked more than 100 attempts to expand midlevel clinicians’ scopes of practice since 2019.
Those barriers may prevent incompetent clinicians from entering the market and thereby protect some patients from low-quality care. That is the ostensible purpose of such laws.
Yet clinician licensing also reduces access to quality care in several ways. First, it increases prices. Licensing increases prices within each profession by increasing the cost of entering that profession. “As you increase the cost of the license to practice medicine you increase the price at which medical service must be sold and you correspondingly decrease the number of people who can afford to buy this medical service.”
Licensing increases prices by requiring patients to obtain services from more-expensive clinicians. Basic primary care generally costs 30 percent less in nurse practitioner–staffed retail clinics than in physicians’ offices. States that prohibit nurse practitioners from practicing independently (see Figure 2) require them to pay up to $15,000 annually to collaborate with a physician, which increases prices for those services. The American Medical Association advocates such restrictions even as it grudgingly admits that midlevel clinicians can provide services within their training at a level of quality comparable to when physicians provide the same services.
Licensing increases prices by prohibiting many health professions outright. Only 14 states allow dental therapists to practice at all (see Figure 3). Patients in the remaining states must see higher-cost dentists for the same services.
Second, licensing blocks access to quality care by reducing the supply of high-quality providers as well as low-quality providers. Licensing may actually reduce the average quality of medical care by inhibiting higher-quality forms of health care delivery.
Licensing blocks free medical care for the poor. The charitable organization Remote Area Medical (RAM) has turned away thousands of patients in need because licensing laws blocked highly qualified volunteer clinicians from around the country from practicing in states where RAM held clinics. “RAM treated 7,000 patients in one week in Los Angeles, but turned away thousands more due to a shortage of California-licensed volunteers.” After a tornado struck Missouri in 2011, RAM “went to Joplin, Mo., with a mobile eyeglass lab. But they were not allowed to make free glasses because their volunteer optometrists and opticians were not licensed in the state.” Licensing often prevents such organizations from even holding clinics at all. RAM’s late founder Stan Brock explained: “We’ve certainly talked to the New York authorities about holding one … in the Bronx.… But again the permission was denied on the licensing issue.” There is no quality-based argument for blocking clinicians with licenses from other states from providing free medical care to the poor.
Licensing blocks access to quality care by reducing the overall supply of clinicians, leaving many patients with no access to care at all. Between 1900 and 1930, shortly after states began controlling entry into the medical profession, the number of physicians per capita fell by 28 percent. One analysis found that “more than a third of 910 small towns that had physicians in 1914 had been abandoned by doctors by 1925.” It was not just low-quality doctors that licensing blocked from the profession. As licensing laws took effect over this period, “the high costs of medical education and more stringent requirements limited the entry of students from the lower and working classes.” Licensing boards closed many medical schools, including five of only seven historically black medical schools. The artificial shortage of medical school slots facilitated discrimination against immigrants, African Americans, women, and Jews in admissions. It should go without saying that preventing these groups from entering the profession has nothing to do with improving quality and instead reduced quality. The legacy of such quality-reducing discrimination persists to this day.
Licensing blocks access to the highest-quality providers in the country, forcing patients to settle for whatever clinicians happen to hold a license in their state. Patients have a right to travel to receive treatment from top specialists at the Cleveland Clinic, Memorial Sloan Kettering Cancer Center, the Mayo Clinic, or other leading medical centers across the country. Licensing denies patients their right to consult those same clinicians via telehealth without leaving home.
Licensing reduces access to high-quality care by blocking entry from integrated, prepaid group plans like Kaiser Permanente. Such systems are strong on dimensions of quality such as coordinating care, conducting comparative-effectiveness research, and offering conveniences like electronic communications, scheduling, and medical records. Many consumers appreciate and embrace this model. Such systems compete on price by making fuller use of midlevel clinicians. Scope-of-practice restrictions disproportionately hinder such systems by depriving them of a key competitive advantage and by requiring them to develop new workflows to conform to each state’s different and ever-changing scope-of-practice rules. Incumbent physicians have even stripped licenses from the physician who founded Kaiser Permanente and others whose only crime was to found or participate in similar plans across the country. The legacy of such discrimination also persists.
States use licensing laws to restrict access to care for reasons that have nothing to do with quality. At least 19 states reduce the supply of clinicians by revoking licenses of clinicians who default on student loans.
Finally, licensing does little to discipline clinicians who actually harm patients. A study by the consumer watchdog Public Citizen found that between 1990 and 2005, “only 33.26 percent of doctors who made 10 or more malpractice payments were disciplined by their state board—meaning two-thirds of doctors in this group of egregious repeat offenders were not disciplined at all.”
Licensing does more to protect the incomes of incumbent clinicians than to protect patients from low-quality care. It adds little if anything to the protections that the medical malpractice liability system and market forces provide. In the absence of clinician licensing, courts would continue to hold individual clinicians and health care organizations accountable for the harm they cause. Hospitals, health plans, and other organizations would continue to evaluate the competence of clinicians via board certification, private credentialing organizations, and their own internal processes.
In the absence of clinician licensing, market forces could provide even greater quality protections. Eliminating licensing would allow greater innovation and competition in health care delivery. Integrated, prepaid group plans could improve quality directly through greater care coordination and health services research. Greater demand for private credentialing and the desire to protect brand names and reputations together would do more than licensing does to safeguard patients from incompetent providers.
Repeal Medical Licensing
Clinician-licensing laws are a mistake that has done enormous harm to patients. Mere tinkering cannot fix them. Government cannot insulate such laws from the influence of incumbent clinicians. Even if it could, government would remain incapable of striking a proper balance between access and safety for millions of patients across billions of medical encounters.
State governments should repeal clinician-licensing laws. At a minimum, states should recognize clinician licenses from other states and other third-party credentialing organizations.
Repealing clinician licensing would reduce the cost of medical care while improving quality. In the absence of licensing, innovators would develop new ways to use midlevel clinicians. Consumers would benefit from greater choice and competition among different delivery and payment systems. Prices would fall for everything from medical education, primary, specialty, and hospital care to health insurance. Repealing licensing would bring health insurance and medical care within reach of many more low-income Americans. It would reduce the number of patients who cannot afford the care they need and reduce the cost of subsidizing those who remain.
Entry by new, higher-quality delivery systems, plus the health-services research and competition they would generate, would improve quality. Such competition would add to the quality assurance mechanisms that would continue to operate in the absence of licensing, including the medical malpractice liability system, board certification, and private credentialing organizations. If repealing clinician licensing is politically infeasible, policymakers must stop licensing laws from acting as a barrier to entry for clinicians licensed by other states.
States must stop licensing from blocking free charitable care for the poor. RAM founder Brock wrote, “In the United States … for some extraordinary reason, practitioners educated and licensed in one state are not allowed to cross state lines to provide free care for needy Americans.” States should enact Good Samaritan laws like those that Connecticut, Illinois, Missouri, and Tennessee pioneered so that clinicians from other states can give away free medical care to the poor. As Brock once testified: “One of the saddest parts of trying to help these people is on the last day of a free RAM event we always have to tell some of them we are sorry, but we cannot see any more patients.… If the government would allow willing volunteer practitioners to cross state lines, fewer people will be turned away.” Volunteer clinicians would still be liable for malpractice under the laws of the patient’s state or the contractual liability rules the patient and clinicians agree to honor.
States must give rural and other patients access to top specialists by recognizing the licenses of telehealth providers in other states. One way to do so is to redefine the location of care from that of the patient to that of the provider—that is, the state where the provider already holds a license.
States can accomplish both of those reforms at once by recognizing clinician licenses from all other states. Arizona has enacted a law that greatly reduces the barriers to out-of-state clinicians practicing in the state.
Congress can use its power under the Commerce Clause to require states to recognize medical licenses issued by other states. In a narrower fashion, Congress can use its power under the Commerce Clause to promote telehealth by redefining the location of the practice of medicine to be that of the clinician.
Medical Facilities
Markets also make medical care better, cheaper, and safer through competition between medical facilities—between retail clinics and physician offices; between urgent care clinics and hospital emergency departments; between standalone imaging centers, radiology practices, and hospital imaging facilities; and between ambulatory surgical centers, specialty hospitals, and general hospitals.
Many states impose laws requiring hospitals, nursing homes, and even physician offices to obtain a “certificate of need” (CON) from a state planning agency before opening or expanding a medical facility or investing in new equipment. CON laws violate the right of patients to choose which medical facilities they patronize. They are a leading barrier to the sort of competition that reduces prices and improves quality.
The rationale for CON laws is that by restraining the supply of hospital beds, the government could restrain medical spending. In 1974, the federal government encouraged states to adopt CON planning.
CON laws failed to slow the growth of medical spending. In a survey of the empirical literature on CON laws, health economist Michael Morrisey writes that those studies “find virtually no cost-containment effects.… If anything, CON programs tended to increase costs.” The failure of CON laws to achieve their stated aims led the federal government to lift its CON-planning mandate in 1987 and also led many states to eliminate their laws. Yet many states have maintained and even expanded their CON requirements.
Nor do CON laws appear to have increased the quality of care. Examining cost and outcomes data for coronary artery bypass grafts, economists Vivian Ho and Meei-Hsiang Ku-Goto found, “CON regulations … may not be justified in terms of either improving quality or controlling cost growth.” Physician-economist Daniel Polsky and colleagues found that laws imposing CON on home-health agencies have “negligible” effects on quality or costs.
Repeal Incumbent-Veto Laws
Perhaps because CON laws have done nothing to contain spending, they have been a boon for incumbent health care providers. Like clinician-licensing laws, CON laws empower incumbents to block new entrants and thereby protect themselves from competition. Morrisey explains:
A reasonably large body of evidence suggests that CON has been used to the benefit of existing hospitals. Prices and costs were higher in the presence of CON, investor-owned hospitals were less likely to enter the market, multihospital systems were less likely to be formed, and hospitals were less likely to be managed under for-profit contract.… The continued existence of CON and, indeed, its reintroduction and expansion despite overwhelming evidence of its ineffectiveness as a cost-control device suggest that something other than the public interest is being sought. The provider self-interest view is worthy of examination.
Indeed, when new entrants apply for certificates of need, incumbent hospitals and other providers object the loudest. Law professor Sallyanne Payton and physician Rhoda M. Powsner explain that although the stated rationale of CON laws is to reduce health care spending, this claim “has diverted attention from the actual economic and political imperatives that led to and presently sustain certificate-of-need regulation. To attribute CON legislation to [cost reduction] is to mistake a convenient theoretical justification for an actual motivation.”
States should eliminate CON laws immediately without any concessions to the inefficient incumbent providers they protect from competition. CON laws harm consumers and taxpayers by increasing health care prices without improving quality. They deny patients their right to choose their medical facilities and the benefits of new forms of health care delivery. There is no justification for them and no place in a market economy for such top-down economic planning.
State officials concerned about runaway health expenditures should reduce or eliminate the government subsidies that fuel such spending. (See “The Tax Treatment of Health Care” and “Medicare.”)
Pharmaceutical Regulation
To market a drug or medical device in the United States, manufacturers must first prove to the satisfaction of the U.S. Food and Drug Administration (FDA) that the product is safe and effective for the indication that will go on the product’s label.
The FDA helps patients when it approves beneficial drugs and blocks harmful drugs. Yet the agency can also harm patients, by either approving harmful drugs (a “Type I error”) or denying approval to beneficial drugs (a “Type II error”). Both Type I and Type II errors can cause suffering and death. Economist Ernst Berndt writes, “A central tradeoff facing the FDA involves balancing its two goals—protecting public health by assuring the safety and efficacy of drugs, and advancing the public health by helping to secure and speed access to new innovations.”
The tradeoff between the number of harmful drugs the FDA approves and the number of beneficial drugs it delays or rejects—that is, between Type I and Type II errors—is unavoidable. Reducing the number of harmful drugs (Type I errors) requires higher standards of evidence, more testing, more time, and more expense. Those measures necessarily increase the number of beneficial drugs the FDA delays or rejects, and they reduce the number of beneficial drugs that manufacturers develop (Type II errors). Conversely, reducing the number of beneficial drugs the FDA delays or rejects (Type II errors) requires easing those barriers to market entry, which inevitably leads to the approval of a greater number of harmful drugs (Type I errors).
As an agency that responds to Congress rather than to patients, the FDA faces an inherent information problem that inevitably leads to unnecessary patient suffering and death. Though Type I and Type II errors can be equally dangerous, Table 1 illustrates a very important difference from the FDA’s perspective. The political system penalizes FDA officials when a patient dies from a harmful drug the officials approved (Type I error). It far less often penalizes agency officials when a patient dies because they blocked or discouraged the development of a beneficial drug (Type II error).
- Type I errors bring swift and certain retribution down on agency officials. The victims are easily identifiable. Patients and the public can easily trace the victims’ injuries to the FDA’s decision. The victims, their loved ones, the media, and Congress can hold FDA officials to account for approving a harmful product. Importantly, FDA officials know Type I errors lead to congressional hearings, public disgrace, and possibly the end of their careers.
- Type II errors bring almost no consequences for FDA officials. Even though delaying or blocking beneficial drugs can harm patients as much as approving unsafe drugs can, it is typically impossible to hold FDA officials to account for Type II errors. Victims of Type II errors are much harder to identify. It appears the disease, not the FDA, killed them. Typically, neither the victims, nor their loved ones, nor FDA officials can identify which patients an unapproved but beneficial drug might have helped. Victims and their families may never have heard of the drug, perhaps because the high cost of avoiding Type I errors deterred companies from ever developing it.
As a result of this fundamental information asymmetry, the political system can discipline FDA officials only when their decisions cause patients to suffer or die from Type I errors. It effectively cannot discipline FDA officials when their decisions cause patients to suffer and die from Type II errors. Dr. Henry Miller, a former FDA official, describes how this information asymmetry affects the decisions of FDA officials:
In the early 1980s, when I headed the team at the FDA that was reviewing the [new drug application] for recombinant human insulin, the first drug made with gene-splicing techniques, we were ready to recommend approval a mere four months after the application was submitted (at a time when the average time for [new drug application] review was more than two and a half years).… My supervisor refused to sign off on the approval—even though he agreed that the data provided compelling evidence of the drug’s safety and effectiveness. “If anything goes wrong,” he argued, “think how bad it will look that we approved the drug so quickly.”… The supervisor was more concerned with not looking bad in case of an unforeseen mishap than with getting an important new product to patients who needed it.
As a result of this information problem and the perverse incentives it creates, the FDA typically tolerates only a 2.5 percent chance of Type I error when determining whether to approve new drugs. Biostatistician Leah Isakov and colleagues estimate that if the agency’s goal is to save lives, it should be much more tolerant of Type I errors. They estimate that for hypertensive disease, the agency should tolerate a 7.6–9.4 percent chance of Type I errors. For cirrhosis of the liver, it should tolerate a 15.3–17.7 percent chance. For pancreatic cancer, it should tolerate as much as a 27.8 percent chance.
Indeed, cost–benefit analyses consistently find that, at the margin, FDA regulation on balance harms patients’ health.
- Economist Mary K. Olson estimates that when additional revenue from user fees enabled the FDA to review drugs more quickly, the health benefits of quicker access to new drugs were roughly 12 times greater than the costs from additional adverse drug reactions. In other words, the FDA was inflicting 12 times as much harm on patients through Type II errors as it was sparing patients by avoiding Type I errors.
- Economist Tomas Philipson and colleagues found that quicker reviews brought significant health benefits but “did not, in fact, have any effect on drug safety.” This finding implies that the FDA will inflict additional deaths due to Type II errors even if doing so produces no reduction in deaths due to Type I errors.
If FDA officials want to promote health, they should regulate less. They should approve new drugs faster and with less evidence of safety and effectiveness.
Unfortunately, this information asymmetry affects more than just the FDA. Despite such research, many in Congress have sought to give the FDA additional powers to reduce Type I errors.
Government-Imposed Prescription Requirements
Congress also empowers the FDA to determine whether consumers must obtain a prescription before accessing certain drugs. Government‐imposed prescription requirements violate the rights of individuals to make their own health decisions. Here again, the agency’s incentives lead it to impose rules that on balance harm rather than protect patients.
The FDA has used its power to impose prescription requirements to steer consumers toward more dangerous drugs. For years, the agency required prescriptions for nonsedating antihistamines while allowing over-the-counter access to sedating antihistamines, a policy that likely caused air- and auto-travel crashes and fatalities. The FDA blocked access to “Plan B” emergency contraception for more than 12 years. FDA-imposed prescription requirements continue to block access to routine‐use oral contraceptives—which are available without prescription in more than 100 countries—and to life‐saving drugs such as naloxone.
Government‐imposed prescription requirements make patients less safe, not more. Economist Sam Peltzman found:
- “Enforcement of prescription regulation increases poisoning mortality by 50 to 100 percent”;
- “No … statistically significant difference in infectious disease mortality between countries that enforce prescription requirements for antibiotics and those that do not”; and
- “[Prescription] regulation did not reduce—indeed, may have increased—poisoning mortality from drug consumption … poisoning mortality is higher, all else remaining the same, in countries that enforce prescription regulation.”
Since “the FDA would instruct firms to remove from their labels any remaining information that might guide lay users of prescription drugs,” economist Peter Temin argued that government-imposed prescription requirements make consumers more vulnerable to harm by making them more ignorant about health and medicines. “Some part of the gap between the drug knowledge of the average doctor and the average consumer is the product of regulation.” Public health professor Julie Donohue notes this power created “a paradoxical situation … in which potentially dangerous prescription drugs were dispensed to consumers with less accompanying information than [over-the-counter] drugs carried.”
A Better Way of Certifying and Monitoring Drugs and Medical Devices
The FDA’s information problem guarantees that the agency will always value some lives more than others and tolerate unnecessary suffering and death. Fortunately, there is a voluntary, market-based alternative that does not suffer from the FDA’s information problem and that respects the right of patients to make their own medical decisions.
Nobel Prize–winning economist Gary Becker advocated eliminating the FDA’s efficacy standard and returning the FDA to the status quo ante 1962, when the FDA had the power only to block drugs it believed to be unsafe. Peltzman argues that even the safety requirement delivers more harm than benefit. Another Nobel Prize–winning economist, Milton Friedman, proposed eliminating the FDA entirely. As long as a government agency exists whose purpose is to protect patients from harmful drugs, it will always focus disproportionately on Type I errors at the expense of overall patient health.
Congress would do better to eliminate any role for the FDA in certifying the safety and efficacy of drugs or in determining which drugs consumers should need prescriptions to purchase.
Eliminating the FDA would increase patient demand for private certification of safety and efficacy, which currently exists only informally. The threat of liability for harmful products would create powerful incentives for pharmaceutical manufacturers to conduct appropriate testing and seek private certification.
Integrated, prepaid group plans like Kaiser Permanente are uniquely capable of performing safety and efficacy certification. When the FDA wanted to determine whether the pain reliever Vioxx (which it had approved) was causing heart attacks, the agency could not conduct that research itself. It turned to Kaiser Permanente of Northern and Southern California. With liberalization of clinician-licensing laws and reforms that allow consumers to control health spending (see “The Tax Treatment of Health Care” and “Medicare”), additional integrated, prepaid plans could enter the market and offer competing safety and efficacy certifications. Different plans would cater to different risk preferences by applying different approval requirements. Each plan’s reputation for quality (and ability to attract enrollees) would depend on the perceived value of its seal of approval. Unlike the FDA, prepaid group plans could consider cost-effectiveness as a criterion for approval. Unlike the FDA, they could closely monitor drug safety and efficacy after approval and could more quickly detect adverse drug reactions. Patients within or outside such plans would rely on whichever plan’s seal of approval fit their own risk preferences.
Market-based certification would save more lives by striking a better balance between Type I and Type II errors. No one would have the power to force patients to suffer Type II errors. Market-based certification respects the freedom of doctors and patients to make treatment decisions according to individual circumstances.
The first step toward reforming the regulation of drugs and medical devices may therefore be to eliminate the barriers that Congress and state legislatures have erected to integrated, prepaid group plans. (See “Health Insurance Regulation,” “The Tax Treatment of Health Care,” and “Medicare.”)
Concurrently, Congress could allow alternative ways of certifying the safety and efficacy of medical products by granting marketing approval to products approved by other countries’ regulatory bodies.
The next step would be to eliminate either the efficacy standard or the FDA entirely. Either would save lives, on balance, because patients would get quicker access to more beneficial new drugs. While patients would also have quicker access to harmful drugs, at least three factors make that unfortunate effect tolerable. First, more patients would live and thrive thanks to greater innovation and quicker access to helpful drugs than would suffer as a result of harmful drugs. Second, eliminating either the efficacy standard or the FDA itself would lead to greater skepticism of new drugs by doctors and patients. Third, innovations by prepaid group plans and others would more quickly detect and stop adverse drug reactions.
Medical Liability Reform
The right to sue health care providers for medical malpractice is a crucial civil right. Individuals are not free to make their own health decisions if health care providers can impose unwanted costs on patients.
The right to sue for medical malpractice is also an important tool for protecting patients from injury due to negligent care. Patients typically have little information about the quality of care. To the extent that the medical malpractice “system” imposes the costs of negligent care on providers, it encourages providers to take steps to improve quality.
Nevertheless, many people in the United States complain—with some justification—that this system performs poorly. “The medical malpractice system is slow, expensive … stressful to both sides, contentious, prone to error in both directions (i.e., payment for weak claims and nonpayment for strong claims), and perceived by everyone involved as inhumane.” According to one estimate, “it costs $1.33 in overhead to deliver $1 to negligently injured plaintiffs.” Even so, research suggests the system does not do enough to discourage negligent care. Physicians and other providers—who see often-dramatic increases in malpractice insurance premiums—have intermittently declared this system to be in “crisis” for more than 30 years.
Scholars have proposed various reforms. California and Texas have limited the amount patients can recover for noneconomic damages to $250,000 per injury. Other proposals include legislative limits on contingency fees for plaintiffs’ attorneys; “no-fault” compensation systems for medical injuries, such as the limited programs adopted in Florida and Virginia; alternative forms of dispute resolution, such as arbitration and special medical courts; the English rule of costs (“loser pays”); and reform of the collateral source rule.
Each of these reforms would leave some patients better off—typically by reducing prices for medical care—at the cost of leaving other patients worse off. “Loser pays” reforms often reallocate the costs of frivolous lawsuits to the correct party. However, that rule deters less affluent patients from seeking legal redress for legitimate grievances. Limits on contingency fees could expand access to medical care by reducing prices, but at the cost of denying compensation to injured patients whose cases plaintiffs’ attorneys deem too expensive to pursue. Perhaps most important, any reduction in provider liability potentially jeopardizes patient safety by reducing the incentives for providers to avoid negligent care.
In particular, caps on damages could expand access to health care by reducing payouts and liability insurance premiums, but at the cost of leaving some injured patients with uncompensated losses. Damage caps in California and Texas force patients to bear the cost of any noneconomic losses they suffer in excess of $250,000.
Moreover, damage caps do not appear to solve the system’s problems or even deliver on the promises of supporters (disproportionately, physicians) that they will increase physician supply or reduce health care spending. A series of empirical studies on Texas’s damage caps concluded:
Texas’s damage cap dramatically reduced the number of medical malpractice cases and total payouts to plaintiffs, with an especially strong effect on elderly plaintiffs. But Texas’s tort reform package had no discernible, favorable impact on broader measures of health system performance. Health care spending growth did not slow, and physician supply did not increase.… While reform strongly benefited providers, the evidence that it had significant benefits for the broader health care system is simply not there.
Like clinician-licensing regulation, much of what physician groups propose with regard to medical malpractice liability benefits physicians at the expense of patients.
Many Republicans want Congress to enact a nationwide set of limits on malpractice liability. The U.S. Constitution does not authorize Congress to impose substantive rules of tort law on the states. Though the federal government may enact technical procedural changes to tort law, state legislatures are the proper venue for correcting excesses in their civil justice systems. The fact that medical professionals can avoid states with inhospitable civil justice systems gives them significant leverage when advocating state-level medical liability reforms and gives states incentives to enact such reforms. Indeed, many states have.
Though state action is preferable to federal action, state-imposed medical malpractice reforms share two flaws with federal reform. First, imposing on all patients and providers any single set of limits on the right to sue for medical malpractice will help some patients but hurt others. Second, though patients should be free to avoid harmful rules, making any single set of rules mandatory and codifying them in statute makes removing harmful rules extremely difficult.
A more patient-friendly and liberty-enhancing approach would allow patients and providers to write their own medical malpractice reforms into legally enforceable contracts. For cases of ordinary negligence, patients could choose the level of protection they desire, rather than have government impose on them a uniform level of protection (and the accompanying price tag). Providers could offer discounts to patients who agree to limits on compensation in the event of an injury. Patients who don’t agree could pay the higher, nondiscounted price or seek a better deal from another provider. Freedom of contract would thus make medical care more affordable to many low-income patients.
Insurance companies could facilitate such contracts on behalf of their enrollees. Those companies would have strong incentives to ensure that such contracts provide adequate protection; otherwise, the insurers could face higher claims from injured patients who could not collect the full extent of their damages.
Regular tort rules would continue to apply in cases where patients and providers could not or did not contract around them, where patients were subject to duress, or where providers were guilty of intentional wrongdoing or reckless behavior.
Freedom of contract would also enhance quality competition. Providers who invest in processes that avoid patient injuries could offer equivalent or more expansive malpractice protections than their competitors at a lower cost. Low-quality providers would not be able to do the same. They would therefore face strong financial incentives to improve quality.
Such contracts are not possible today because courts have invalidated them as “contracts of adhesion” or “against public policy.” The courts’ refusal to honor those contracts restricts the freedom of adults to make mutually beneficial exchanges that hurt no one else. It also increases the cost of providing medical care to the poor, which has undoubtedly reduced their access to care.
To remedy this undue and costly restriction on liberty, courts should abandon their current policy and enforce contractual limitations on the right to sue for medical malpractice. If courts refuse, state legislatures should require them to do so. Nobel Prize–winning economist Richard Thaler and law professor Cass Sunstein write:
In our view, state lawmakers should think seriously about increasing freedom of contract in the domain of medical malpractice, if only to see whether such experiments would reduce the cost of health care without decreasing its quality. Increasing contractual freedom won’t solve the health care crisis. But it might well help—and in this domain every little bit of help counts.
The medical malpractice system does a poor job of providing relief to injured patients, preventing frivolous lawsuits, or discouraging negligence. The remedies for these shortcomings are not obvious. A dynamic marketplace that allows parties to experiment with—and abandon—different malpractice rules is the quickest and surest way to find those solutions.
Suggested Readings
Black, Bernard S., et al. Medical Malpractice Litigation: How It Works, Why Tort Reform Hasn’t Helped. Washington: Cato Institute, 2021.
Cannon, Michael F. “Health Care’s Future Is So Bright, I Gotta Wear Shades.” Willamette Law Review 51, no. 4 (2015): 559–71.
Epstein, Richard A. “Medical Malpractice: The Case for Contract.” American Bar Foundation Research Journal 87, no. 1 (1976): 87–149.
Friedman, Milton. “Occupational Licensure.” In Capitalism and Freedom. Chicago: University of Chicago Press, 1962, chap. 9.
Singer, Jeffrey A., and Michael F. Cannon. “Drug Reformation: End Government’s Power to Require Prescriptions.” Cato Institute White Paper, October 20, 2020.
Svorny, Shirley. “Could Mandatory Caps on Medical Malpractice Damages Harm Consumers?” Cato Institute Policy Analysis no. 685, October 20, 2011.
———. “Medical Licensing: An Obstacle to Affordable, Quality Care.” Cato Institute Policy Analysis no. 621, September 17, 2008.
Svorny, Shirley, and Michael F. Cannon. “Health Care Workforce Reform: COVID-19 Spotlights Need for Changes to Clinician Licensing.” Cato Institute Policy Analysis no. 899, August 4, 2020.

About the Author


This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.